
Page 42 - 《中国药科大学学报》2026年第1期
P. 42
36 学报 Journal of China Pharmaceutical University 2026, 57(1): 34 − 45 第 57 卷
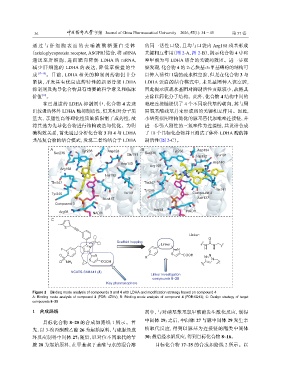
通 过 与 肝 细 胞 表 面 的 去 唾 液 酸 糖 蛋 白 受 体 的同一活性口袋,且均与口袋内 Arg168 残基形成
(asialoglycoprotein receptor, ASGPR)结合,将 siRNA 关键相互作用(图 2-A,图 2-B),揭示化合物 4 中噻
递送至肝细胞,进而靶向降解 LDHA 的 mRNA, 唑甲酸为与 LDHA 结合的关键药效团。进一步观
减少肝细胞的 LDHA 的表达,降低草酸盐的生 察发现,化合物 4 的 2-乙炔基-5-甲基噻吩的结构可
成 [17−18] 。目前,LDHA 相关的抑制剂药物仍十分 以伸入活性口袋的疏水性空腔,但是在化合物 3 与
紧缺,开发具有优良成药特性的新型骨架 LDHA LDHA 蛋白的结合模式中,未见基团伸入该空腔,
抑制剂及先导化合物具有重要的科学意义和临床 因此提示该疏水基团对抑制活性贡献较小,故将其
价值 。 去除以简化分子结构。此外,化合物 4 结构中间的
[19]
在已报道的 LDHA 抑制剂中,化合物 4 表现 吡唑连接链提供了 4 个不同取代基的朝向,其与周
出较强的体外 LDHA 酶抑制活性,但其相对分子质 围氨基酸残基并未形成强的关键相互作用。因此,
量大、亲脂性高等理化性质缺陷限制了成药性,故 本研究拟用结构简化的脲基替代原吡唑连接链,并
将其选为先导化合物进行结构改造与优化。为明 进一步引入刚性的三氮唑作为连接链,共设计合成
确构效关系,首先通过分析化合物 3 和 4 与 LDHA 了 18 个目标化合物并且测试了体外 LDHA 酶的抑
共晶复合物的结合模式,发现二者均结合于 LDHA 制活性(图 2-C)。
A Tyr238 Asp194 B Tyr238 Asp194
Ser236 Glu191 Ser236 His192 Glu191
Asp140 Arg168 Asp140
Arg168
His192
Thr247 Thr247
Tyr246 Ile141
Tyr246 Ile141 Compound 4
Asn137 Asn137
Compound 3
Arg98 NADH Arg98 NADH
C
S
Linker:
O
Scaffold hopping Linker
N N N
F N N H H
O N S COOH N N
S S R N
O NH 2 COOH
NCATS-SM1441 (4)
Linker investigation
compounds 8−25
Key pharmacophore
Figure 2 Binding mode analysis of compounds 3 and 4 with LDHA and modification strategy based on compound 4
A: Binding mode analysis of compound 3 (PDB: 4ZVV); B: Binding mode analysis of compound 4 (PDB:6Q13); C: Design strategy of target
compounds 8−25
1 合成路线 剂中,与对硝基苯基氯甲酸酯发生酰化反应,制得
目标化合物 8~25 的合成如路线 1 所示。首 中间体 29;之后,中间体 27 与脲中间体 29 发生亲
先,以 3-溴丙酮酸乙酯 26 为起始原料,与硫脲经成 核取代反应,得到以脲基为连接链的酯类中间体
环反应制得中间体 27;随后,以对位不同取代的苄 30;最后经水解反应,得到目标化合物 8~16。
胺 28 为起始原料,在甲基叔丁基醚与水的混合溶 目标化合物 17~25 的合成如路线 2 所示。以